1. Forcite 계산 시작하기
- 현재 모델은 최적화 된 것이 아님
- 따라서 최적화 시키기 위하여 forcite 모듈 이용

- forcite - calculation 선택
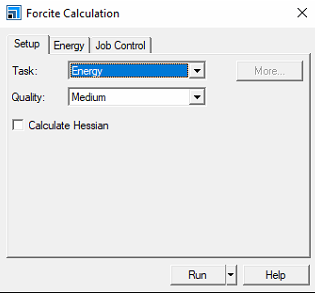
- 창 생성
- Task는 DMol3와 거의 유사함
1) Setup
- energy : 싱글 포인트의 에너지
- dynamic : DMol은 전자를 움직이면서 온도나 압력을 넣을 수 있음. 그러나 전자가 이동하는 것 까지 다 계산을 해야 하기 때문에 계산 시간과 양이 매우 큼
: Forcite의 경우, 아톰마다 본딩해있는 결합 정보가 다 저장되어 있기 때문에, 계산 양이 많이 필요하지 않음
: 따라서 보통 MM을 이용해서 계산해야 함
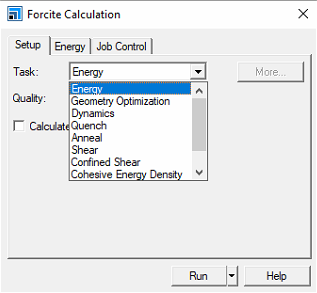
- Quanchi : 온도를 차츰 내려가면서 폴리머의 안정된 구조를 찾는 기능
- Anneal : 온도를 높였다가 낮췄다가를 반복함으로써 안정된 구조를 찾는 기능

- 우리가 현재 하는 것은 안정화(최적화)된 구조를 찾는 것이므로 Geometry Optimization을 선택
- More 버튼을 눌러 상세 조건을 설정함

- Algorithm의 경우, Smart 설정 : 1차 미분, 2차 미분에 대해서 안정된 구조를 찾아낼 수 있도록 혼합된 기능을 가지는 smart 옵션을 사용하는 것
- External pressure : 외부 압력을 지정할 수 있음. 필요할 경우에 설정
- Optimize cell을 하지 않을 것 (이전에 amophous cell을 계산할 때 처럼)
- keep motion group : 분자나 폴리머의 움직임을 제한하고 싶을 때 사용
2) Energy Task

- 중요한 것은 forcefield임
- forcefield의 종류는 매우 많은데, 그 중에서 COMPASS와 Universal를 비교 할 것
- COMPASS3를 체크하고 more버튼 클릭

- 원소에 따라서 어떠한 원소인지 다 표시하고 있고, 정보가 저장되어 있음
- 하단의 체크를 해제하면, 지금 내 스케치에 어떤 원소가 있는지 정보가 나타남
- COMPASS를 사용하기 전엔, 사용자가 자신의 스케치를 보고 원소를 하나하나 넣어야 했는데, COMPASS는 자동으로 정보가 나오니까 매우 편리함
- 그만큼 forcefield를 제대로 설정하지 않으면 정확한 계산 값을 얻지 못하고, 내 스케치의 원소와는 다른 원소를 지정하게 되면 계산에 실패하게 됨
- 따라서 MM 같은 경우 제대로 된 forcefield 설정(제대로된 원소 정의)을 하는 것이 매우 중요
- universal의 경우, 정의되는 원소가 매우 대략적임 (자세하지 않음)
- 또한 MM은 Charge에 대해서도 별다른 설정을 해야 함
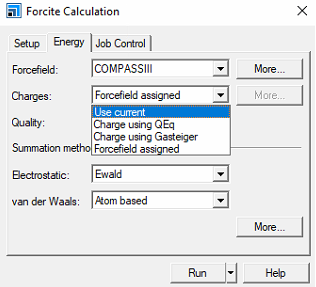
- Forcefield assigned : 최근 forcefield에 대해서는 아톰에 따라서 차지 값을 계산해놨음
- 반면에, 본인이 양자 계산을 통해 차지 값을 정확히 찾아낸 경우에는 자동적으로 계산 해 놓은 것이 아닌 use current를 설정하는 것이 나음

- bondinteraction : 본드 결합 정보를 계산
- 아톰을 하나 기준으로 본드가 세개 이상인 것은 nonbond로 설정
- 튜토리얼에서 진행한 것 처럼 Group based로 설정
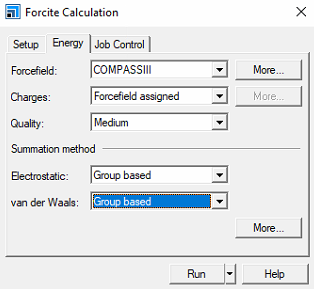
2. 결과 해석하기

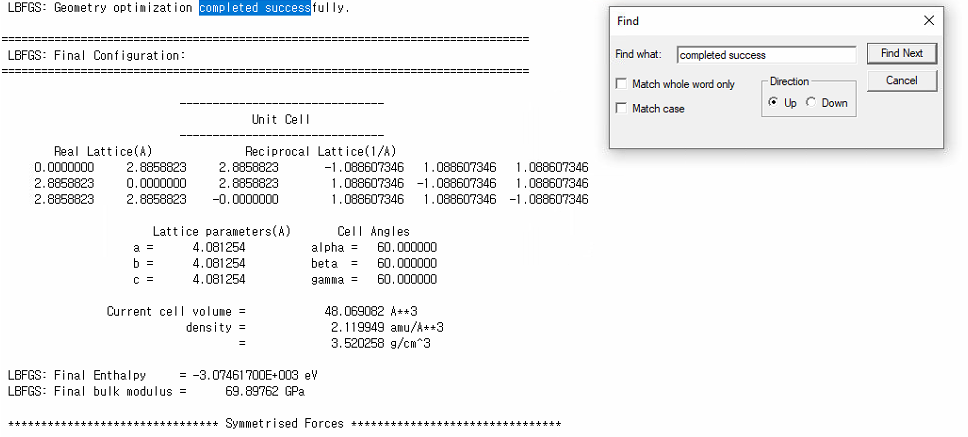
- 계산이 성공적으로 된 것을 알 수 있음
1) output (cell.txt)

- total enthalpy : valence energy(아톰이 갖고 있는 + cross terms + non-bond energy 이 세개의 에너지의 합으로 이 에너지가 결정됨
- 따라서 양자 계산과의 에너지 용어에 대해서는 구분을 해야 함
- 이 에너지 값을 양자 계산에 넣어서 직접적인 비교를 할 수 없음

- 초기 에너지는 매우 불안정한 값

- 나중 에너지는 최적화가 진행되어 매우 안정한 것을 알 수 있음
① valence energy
- 폴리머 기준으로 예를 들었을 때, 본드가 세개인 것에 대한 아톰의 에너지
- 즉

- 이 값만 계산한 에너지
- 이 세개 이외의 나머지 본드에 대해서는 non-bond energy 값임
② non-bond energy

③ valence energy (cross term)

- 최종적으로 계산이 잘 진행됐는지 확인하기 위해
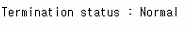
- 정상적으로 계산이 됐음을 알 수 있음
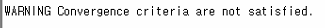
- 처음에 만든 구조가 임시적인 구조였기 때문에, 이 경고창이 뜨지 않도록 여러 번을 반복해줘야 함
- 반복작업을 하기 위해서 주의해야 할 점은 처음의 cell의 파일로 하면 안되고, 한 번 최적화가 된 cell 파일을 열어줘야 함
- 똑같은 조건으로 계산하기 위해서 cell calculation을 클릭하면 됨
- 우리가 안정된 구조를 찾는 이유? 불안정한 구조로 dynamics task를 하게 되면 계산이 fail이 될 확률이 높음
3. 한 번 더 최적화 하기

- 차트가 계속해서 내려가는데, 불안정한 부분이 보임
- 따라서 최적화를 계속 할 필요가 있음

- 최적화를 더 할 필요가 있음을 output 파일을 통해서도 확인할 수 있음
4. Forcite의 모듈에서 Aneal task 진행하기


- Aneal을 선택
- Aneal은 앞서 언급했듯이, 온도를 높였다 낮췄다를 반복하면서 (사인 그래프 형식) 안정화된 구조를 찾는 계산
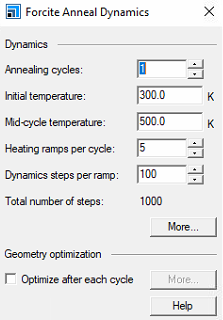
- 위와 같이 cycle, 온도 설정

- Energy 창을 다음과 같이 설정
- 항상 주의해야 할 점은 마지막에 최적화된 파일로 계산을 진행해야 한다는 것

- 계산이 완료 되었음
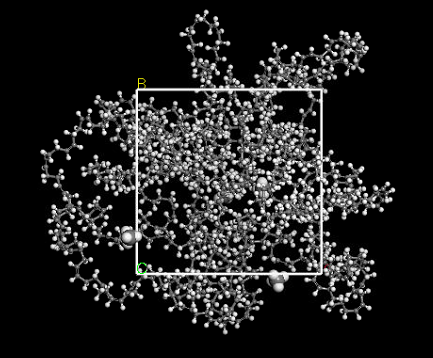
- Aneal의 목적은 안정적인 구조를 찾는 것이므로 cell.xsd 파일을 열어서 안정적인 구조를 확인
5. Dynamics 계산
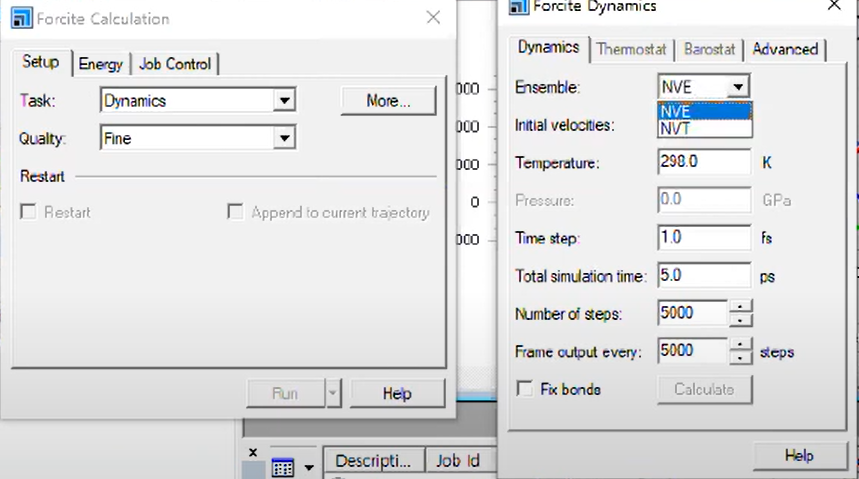
- cell.xsd 파일을 활성화 한 뒤에, calculation 활성화
- Dynamcics task는 more 버튼을 보면 앙상블을 선택할 수 있음
- N : 아톰의 수가 고정되어 있음
- V : 부피를 고정하고 싶을 때, E : 에너지, T : 온도
- P : 압력, H : 엔탈피
- 일반적으로 온도나 압력으로 수렴시킨 다음에, 최종적으로 NVE, 즉 에너지를 수렴시킴
" 이 온도, 이런 압력을 받았을 때, 이 구조는 이런 에너지를 가질 것이다."
- dynamics는 오랜 시간이 걸리기 때문에 인내심이 필요
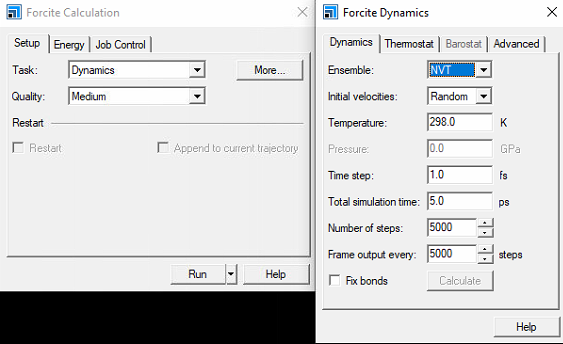
- 앙상블은 NVT로 먼저 온도에 따른 부피의 수렴값을 알아 볼 것임
- Initial velocities : dynamics 계산은 F = ma, 즉 운동 방정식에 기반을 하기 때문에 임의의 속도 값을 지정했을 때, 그 때의 a값이나 F값을 계산해서 그 다음 위치값을 예측함. 따라서 초기 속도를 랜덤하게 주겠다는 얘기

- 위 설정으로 함
- 컴퓨터 사양이 좋지 않기 때문에 5ps로 정함 (마력 단위, 일률이나 동력)
- Frame output every : 몇 스텝마다 캡쳐본을 찍을거냐? (아웃풋을 만들어낼거냐)
- 따라서 3개의 아웃풋 (초기, 그리고 2번)이 나올 것

- Thermal 같은 경우, 내가 온도를 어떤 method를 통해 300K에 수렴시킬 것인지?
- 위키피디아에 자세히 나왔으니 그것을 참고
- 온도 조절 장치라고 보면 됨

- 계산이 성공적으로 완료됐음

- .xtd 파일을 열어서 애니메이션 버튼이 있는지 확인
6. MSD 계산하기

- 지금 폴리머와 메탄이 결합된 구조에서 메탄 분자만 찾아내야 함
- 초기에 스케치 했던 methane.xsd 파일 클릭
- 또 최종 파일인 cell.xtd 파일을 활성화
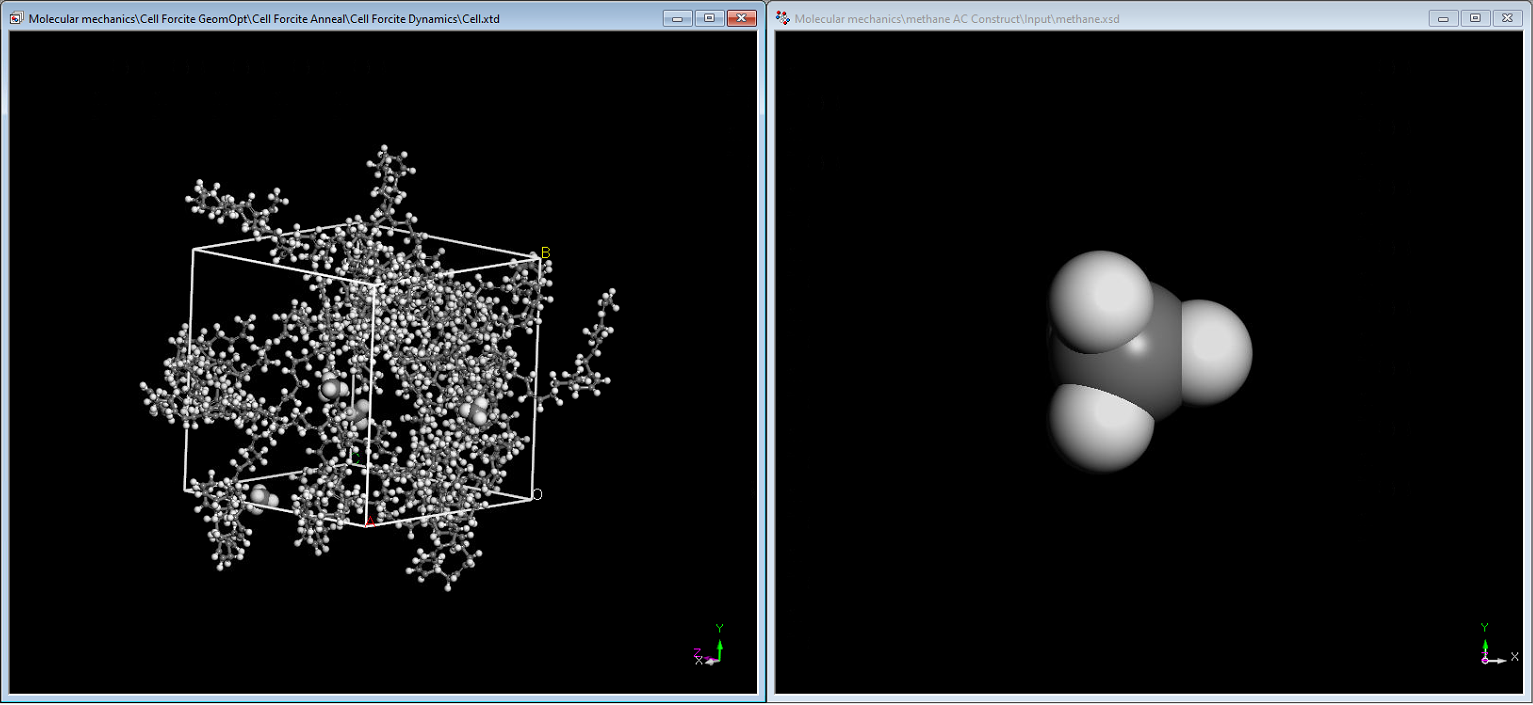
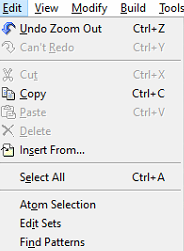
- 메탄 창을 누른 상태에서 edit sets 버튼 클릭

- set이 정의가 안 되어있기 때문에 edit sets 아래에 있는 find patterns 클릭
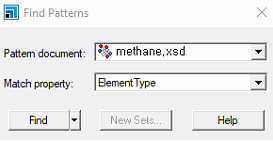
- 위와 같이 설정하고, 최종 파일이었던 cell.xtd 파일을 클릭하면 find 버튼이 활성화 됨

- 자동으로 메탄 분자를 찾아주었음
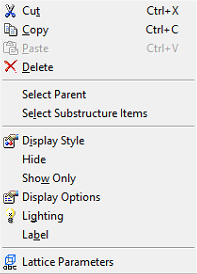
- 여기서 우클릭을 누르고 select substructure items 를 클릭

- 그럼 찾아준 분자를 자동으로 선택해주었음

- 그리고 나서 new sets 클릭
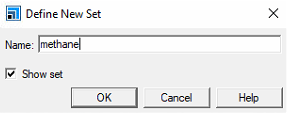
- 이름을 methane으로 정의
- 추가된 것을 알 수 있음
- 애니메이션 버튼을 누르면 다음과 같이 움직임을 알 수 있음
- Forcite 모듈의 analysis 클릭

- 오른쪽 카테고리의 의미 : 분자 구조만 있어도 계산할 수 있음 / dynamics task를 진행해야 계산할 수 있음
- sets는 아까 정의했던 methane을 찾기
- 그러나 사실 현재는 시간 설정이 매우 짧음
- 또 충분히 수렴을 시켜서, 정말 안정한 구조에서 이 분석을 하는게 맞음
- 그래야만이 정말 정확한 값을 얻을 수 있음

- 이 기울기 그래프를 가지고 확산성 값을 알 수 있음
- 이 MSD값
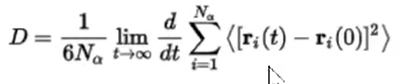
- 이 수식을 자동으로 다 계산해서 얻은 기울기임
- 어떤 용도로 사용하냐에 따라 다르겠지만, MSD는 가장 많이 사용하는 모듈임
'Material Studio > 1. 기본 step' 카테고리의 다른 글
| [계산 영역 및 모듈 정리] (0) | 2021.07.25 |
|---|---|
| 5. Molecular mechanics 모듈 이용하기 - 1 (0) | 2021.07.19 |
| 4. CASTEP을 이용해 계산하기 - 2 (0) | 2021.07.19 |
| 4. CASTEP을 이용해 계산하기 - 1 (0) | 2021.07.19 |
| 3. DMol3를 이용해 계산 진행하기 - 2 (0) | 2021.07.19 |